Copy to clipboard
Copy to clipboard 
Months ago, the idea came to me to write three articles about the lessons learned from metal-on-metal (MoM) implants. My intent was to have three authors write about three important aspects of designing orthopaedic implants: verification test methods, risk analysis and post-market surveillance.
Jean Bigoney, Ph.D., a metallurgist and regulatory consultant, agreed to write about the verification test methods. I was writing about risk analysis, and was asking a Notified Body to write about post-market surveillance. “Professor Jean” was kind enough to help me with the first article, and she was able to get additional information from another scientist in Germany who has conducted a number of wear tests on MoM hip implants for OEMs. I thought enlisting her help was going to be the hard part. I soon learned an unanticipated lesson about MoM implants in the process of writing this article.
The first Notified Body I contacted refused to respond to my request. It wasn’t just a case of overlooking the email. My friend responded to all my emails—except the one about these articles. The second Notified Body I contacted responded by saying, “[NB#2] must decline at this time.” The third Notified Body I contacted responded with a phone call. This person said that [NB#3] did not have any clients with approved MoM implants, and it would be a great opportunity to show how other Notified Bodies should have done things differently. However, this person also said that [NB#3] could not write the article. What makes one Notified Body look bad makes all the Notified Bodies look bad.
In January, the FDA was the latest regulatory body to take investigative action on MoM implants. While assembling a committee to review clinical data and the benefits and risks of MoM hip implants, the FDA developed safety communication recommendations and proposed orders to require OEMs to submit premarket approval applications.
What follows is our two-part primer on metal implant test method results and risk analysis steps OEMs should consider as fallout continues over the use of MoM implants.
A Titanium Metallurgist’s Perspective on MoM
By Jean Bigoney, Ph.D.
“Titanium hip implants? Not in MY patients!” This was the pronouncement of my friend and colleague in Munich, orthopaedic surgeon Dr. Reiner Gradinger, which left no doubt as to his material of choice for his patients. As far as he was concerned, the risk of aseptic loosening from wear debris was too great. Titanium may be widely considered “biocompatible” and exhibit suitable strength in applications as endoprostheses, but titanium was notorious for having poor wear resistance. He wanted no part of it. Cobalt-based implants were, in his mind, safer due to the naturally superior wear resistance.
He wasn’t the only one. As long as surgeons performing revision surgery were seeing evidence of “wear disease,” which they sometimes called “black death” to vividly describe the metallosis of periprosthetic tissue when retrieving cemented titanium prostheses, many remained skeptical about titanium. Aseptic loosening remains the most frequent cause of revision surgery in total hip replacement (THR), according to the Swedish Hip Arthroplasty Register, and prevalence has remained unchanged over the years at roughly 70%. Along with titanium, wear debris from Ultra-high-molecular-weight polyethylene (UHMWPE) has also implicated as a cause of aseptic loosening in hip implants. Therefore, scientists have put a lot of time and effort into studying particles during the last 20 years.
Pathologists looked at the discolored tissue retrieved from revision surgery, and told us that it contained metallic particles about 0.2 mm in size. See Exhibit 1.
Cell biologists, wanting to understand a causal relationship between the particles and osteolysis, looked at particles and how they influence cellular reactions. After all, the fact that metal particles are observed surrounding loose prostheses doesn’t necessarily mean those particles were the cause. This resulted in a slew of in vitro studies done on particles of UHMWPE, titanium and cobalt. Unfortunately, realistic wear debris is hard to come by.
Many investigators resorted to using metal powder intended for sintering parts, but these particles are much larger and much rounder than the small irregular wear particles. Some researchers decided that titanium oxide was a good substitute for the hard-to-get submicron titanium particles. It oxidizes anyway, right? See Exhibit 2.
Conclusions drawn from investigations on easy-to-get particles, as opposed to realistic particles, have limited applicability in explaining what havoc real wear particles might be wreaking in the body. A landmark study in 1993 comparing real wear debris ranging in size from 0.5-3 mm from both Ti-6Al-4V and Co-Cr concluded that while cobalt particles were more toxic, titanium particles were more likely to contribute to aseptic loosening.
We had our marching orders. We assigned the highest risk to aseptic loosening, and particles were to blame, particularly if they were titanium. Knowing what to be on the lookout for, testing labs took over the task of examining particles from wear testing, duly counting them and recording size and aspect ratio according to ASTM F1877.

Exhibit 1: Tissue retrieved from patients Top: tissue samples; bottom: histological image showing wear debris as irregular dark spots. Image originally published in (W. Mittelmeier, E. Steinhauser, M. Schmitt, A. Eichbichler, J. Gregory, H. Rechl, R. Gradinger: Current Aspects of Failure Analysis of Endoprotheses for Joint Replacement (in German) Med. Orth. Tech. 120 (2000) 23-27.
It came as no surprise that if we were going to use metal on metal, it would be cobalt instead of titanium. After all, we knew that cobalt was “wear resistant,” and unlike titanium, is allowed by ISO 21534 on articulating surfaces. And we knew to look at wear debris thanks to the huge body of information on hip replacements.
Cobalt and chrome are not known carcinogens, and we certainly didn’t expect anodic dissolution of a corrosion-resistant alloy. Orthopaedic surgeons were advised to make patients aware of the potential unknowns regarding the possibility of metal ions being released.
What wasn’t supposed to happen was what became known as arthroprosthetic cobaltism. Reports started coming in that patients fitted with MoM hip prostheses were exhibiting symptoms of neurological, cardiac and endocrine disorders. Cobalt wasn’t supposed to leach out of the surface, but evidently it was, possibly exacerbated by being released as particles with a high surface to volume ratio. The risk may not have been properly assessed or tested appropriately.
As of this writing, FDA is only recommending serum testing for metal ions when patients with MoM prostheses develop symptoms indicating that there might be a problem.
Biocompatibility testing on metallic implants is usually performed on liquid extracts collected for a short time, but not on wear debris. With all the wear and fatigue testing that was done to evaluate new implant designs, scientists should consider collecting the serum solution (a liquid used to simulate body fluids) throughout wear and fatigue testing for elevated levels of Co or Cr. The Ringer’s solution could also be used for biocompatibility testing. Currently there is no accepted testing standard to accommodate such testing, although published work using a simple toxicity screening tool has shown collected solution to have toxic effects.
Other test method modifications that might be reconsidered include evaluation of the effect of increased protein levels in the testing solution, and the effect of bone fragments that may remain after surgery on wear behavior.
If titanium implants result in aseptic loosening, and MoM implants cause systemic problems due to leaching of cobalt, is it time for the industry to take another look at ceramic implants? Clinical performance has been promising. However, while ceramic enjoys a reputation for being wear-resistant, there has also been a hesitancy to use it owing to its brittle nature. The risk of catastrophic failure appears to be what is holding us back.
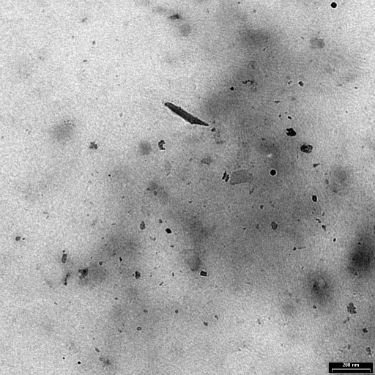
Exhibit 2: Wear particles generated in vitro from an electrolytically polished surface of Ti-6Al-4V ELI following 2000 cycles.
Just before I entered the Orthopaedic Clinic Rummelsberg, as an observer to watch revision surgeries on hips and knees, Chief Surgeon Dr. Günther Zeiler showed me the rack where retrieved prostheses were deposited. “Here, take a look at this,” he said, pulling out a ceramic-on-ceramic ball and socket joint. “Ceramic isn’t supposed to wear, right? So what do you call this?” he asked, pointing to the black powder on the surface. “Sometimes things happen in the body that we can’t reproduce in bench tests.”
In the case of novel material technologies, there is a need for performing pre-clinical studies in animal models to assess the suitability of the material in a long-term study with follow-up after the natural death of the animal. This may be the only way to predict failures caused by a combination of wear and exposure to the environment of the body, and assign risks accordingly.
Helpful information and images were kindly provided by Christian Kaddick of EndoLab Mechanical Engineering GmbH, www.endolab.org.
A Risk Analysis of MoM Implants
By Rob Packard
If you took my class on ISO 14971, the risk management standard for medical devices, I would remind you that you should assume that your device is made to specifications when you are estimating risks for a design risk analysis (e.g. Design Failure Mode and Effects Analysis). For permanent implants, however, the risk analysis should consider the risks posed by new implants that are made to specifications: worn implant and wear particles. Now that you have the benefit of hindsight, you have a new harm to add: arthroprosthetic cobaltism (i.e. systemic cobalt poisoning). The next step is to estimate the probability of occurrence of that harm. As more data is gathered on the performance of MoM implants, there will be estimates published in literature.
The problem with estimating the probability of occurrence from literature is that it is not proactively estimating the risks. If you can’t estimate the probability of occurrence of harm, your company should be developing a post-market clinical follow-up (PMCF) plan to gather this clinical data. One approach would be to create a patient registry and execute a PMCF, which includes periodic testing for metal ions along with imaging of the patient at each time point. This would identify the patients who are exhibiting the greatest exposure to the metal ions, when it begins to occur, and the generation of metal ions can be correlated with evidence of aseptic loosening observed in patient imaging data. The current approach of testing serum levels after the patient exhibits symptoms provides far less clinical data that would act as an early warning system.
Professor Jean proposed a second method for estimating this risk. The serum solution that is used to simulate synovial fluids during in vitro wear testing of implants could be periodically sampled and monitored for the metal ion concentration. She also suggested conducting in vivo pre-clinical testing of implants. These survival studies in animals should include the same periodic testing for metal ions and collection of imaging data that is proposed for PMCF studies. The collection of both in vivo pre-clinical data and PMCF data using similar protocols is critical in establishing potential correlation models between the performance in animal models and human patients.
The above suggestions for estimating the probability of occurrence are expensive, time consuming and were considered negligible risks for cobalt chrome implants—until MoM implants were commercially used for several years. The 2009 version of EN ISO 14971 indicates that an OEM may discard negligible risks in Annex D 8.2. Annex I, Sections 1 and 2 of the Medical Device Directive (MDD), however, requires that all risks need to be reduced as much as possible and need to be balanced, together with all other risks, against the benefit of the device. This is the first point in Annex ZA of the 2012 version of EN ISO 14971.¹
Annex D, section 8 of EN ISO 14971:2009 also indicates that an OEM may reduce risks to a level that is “as low as reasonably practicable” (ALARP concept). This is not consistent with the Annex I, Section 2 of the MDD which requires reducing risk “as far as possible” without there being room for economic considerations. It is quite possible that the European Commission will implement more stringent Common Technical Specifications for resurfacing implants to replace the existing ASTM test methods to ensure that risks are reduced “as far as possible.”
Post-Market Surveillance
Last year, Dr. Hamish Forster at BSI wrote a white paper titled, “The Post-Market Priority: Understanding and Meeting Demand for Effective Post-Market Clinical Follow-Up.”² This article refers to the requirement in Annex X, Section 1.1c for conducting PMCF—or justifying why it is not required. The article also refers to the new guidance document about PMCF, MEDDEV 2.12-2 rev 2, released at the beginning of 2012.
Most OEMs include a justification for why PMCF is not required. In fact, I have reviewed more than 200 technical files and Design Dossiers personally, and I have not seen a single PMCF plan yet. I have seen a few post-market surveillance plans that were titled “PMCF,” but none of the plans met the requirements of MEDDEV 2.12-2 rev 2.
The guidance document is not hard to understand, but compliance with it is expensive. Therefore, every company will have a bias toward justification for no PMCF. Instead of waiting for Notified Bodies and the European Commission to force the issue, I recommend a simple test, which all OEMs can practice.
In Forster’s article, starting at the bottom of the first page, he lists 17 “circumstances under which PMCF may be necessary.” In order to decide if PMCF is necessary for your devices, review Dr. Forster’s article to determine how many of the circumstances apply. Although just one is enough to warrant a PMCF study, if three or more apply to your device there may be no justification for not performing PMCF. In the case of MoM implants, there are seven bullets that apply:
- Innovation: the device’s design, materials, substances, principles of operation, technology or medical indications are novel
- High product-related risk
- Unanswered questions of long-term safety and performance
- Identification of previously unstudied subpopulations which may exhibit different benefit/risk-ratio (e.g., hip implants in different ethnic populations)
- Continued validation in cases of discrepancy between reasonable pre-market follow-up time scales and the expected life of the product
- Verification of safety and performance of device when exposed to a larger and more varied population of clinical users
- Emergence of new information on safety or performance
There may be no scientific basis for the “rule of three,” but a strong PMCF study would have quantified the risk of harm caused by MoM implants much earlier than waiting for the receipt of adverse event reports.
In the future, OEMs should expect the requirement for PMCF studies to be enforced by Notified Bodies. The EU Commission released proposed regulations on September 26, 2012. In that proposal, the phrase post-market clinical follow-up appeared 17 times. In Article 8, the proposal specifically states that a PMCF plan shall be part of the Post-Market Surveillance Plan. In Annex II, Section 6.1d), the proposal states that Technical Documentation shall include the PMCF plan and PMCF evaluation report in accordance with Part B of Annex VIII. Annex VIII even includes PMCF in the title.
A Culture of Openness
The final lesson you need to learn is that you must take every measure to ensure that every member of your design team feels comfortable voicing their concerns and saying that there are problems—even if top management does not want to hear about problems. The failure of regulators to be more critical of MoM implants is a systemic problem that new regulations will attempt to address. However, the root cause is not a lack of regulations. The root cause is a culture that feels pressured to approve new technology, at companies and at regulators, with unknown risks. A similar culture existed at NASA in the 1980s, and it resulted in a decision that caused the deaths of seven astronauts.
Robert Packard of Packard Consulting is a regulatory consultant with 20 years of experience developing products and managing projects in the medical device, biotechnology, pharmaceutical industries. His experience includes research, product development, operations management, manufacturing engineering, equipment design, regulatory affairs, quality assurance and fund-raising. Rob’s passion is training others. He may be reached at [email protected].
Jean Bigoney is a metallurgical and regulatory consultant with 30 years of experience in R&D of materials for aerospace and biomedical implants. She has authored over 50 scientific publications and four book chapters, and was the editor of a book on surface treatment of titanium. After spending most of her career in Germany, she returned to the U.S. in 2002 to start Springfield Metallurgical Services, Inc. (www.smslab.net). Dr. Bigoney also maintains Nu Device Consulting, LLC (www.nu-device.com), focusing on 510(k) submissions for domestic and international companies. She may be reached at [email protected].